Watch Lisa Nodzon, PhD, ARNP, AOCNP, discuss the clinical guidance and how dose modifications for ARs with IMBRUVICA® may help your appropriate patients continue treatment. Discontinuation may be appropriate for some patients.

Dose modifications can help optimize patient management to enable continued treatment when appropriate
Dose modifications for adverse reactions1*
If an AR listed here occurs, interrupt IMBRUVICA® therapy at each occurrence of the same AR.
Once the AR has improved to Grade 1 or baseline, follow the recommended dose modifications.1
Start at approved dose 420 mg
Once daily until disease progression or unacceptable toxicity
ADVERSE REACTION†‡
occurrence
1ST
2ND
3RD
GRADE 3 or 4: other non-hematological toxicities§
Restart at280 mg‖ daily
Restart at140 mg‖ daily
Discontinue
GRADE 3 or 4: neutropenia with infection or fever
Restart at280 mg‖ daily
Restart at140 mg‖ daily
Discontinue
GRADE 4: hematological toxicities
Restart at280 mg‖ daily
Restart at140 mg‖ daily
Discontinue
GRADE 2: cardiac failure
Restart at280 mg‖ daily
Restart at140 mg‖ daily
Discontinue
GRADE 3: cardiac arrhythmias
Restart at280 mg‖ daily
Discontinue
GRADE 3 or 4: cardiac failure
Discontinue
GRADE 4: cardiac arrhythmias
Discontinue
Certain types and severity of cardiac ARs require discontinuation after first occurrence for only IMBRUVICA®.
†See full Prescribing Information for Warnings and Precautions.
‡Grading based on National Cancer Institute-Common Terminology Criteria for Adverse Events (NCI-CTCAE) criteria, or International Workshop on Chronic Lymphocytic Leukemia (iwCLL) criteria for hematological toxicities in CLL/SLL.
§For Grade 4 non-hematological toxicities, evaluate the benefit-risk before resuming treatment.
‖Evaluate the benefit-risk before resuming treatment.
For use with CYP3A inhibitors and inducers, and in patients with hepatic impairment, please see the full Prescribing Information. Consider the risks and benefits of anticoagulant or antiplatelet therapy when co-administered with IMBRUVICA®. Monitor for signs and symptoms of bleeding. Consider the benefit-risk of withholding IMBRUVICA® for at least 3 to 7 days pre- and post-surgery depending upon the type of surgery and risk of bleeding.

Eddie discussed his side effects with his doctor, who decided to reduce his dose.
We were able
to reduce my dosage...and it helped with my side effects.”
Eddie, an IMBRUVICA® patient living with CLL
Patient experiences may vary.
NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines®)
Guidance on ibrutinib (IMBRUVICA®) Dose Modifications for Certain ARs2
In patients with no intolerance, cBTKi can be continued until disease progression, while following recommended dose modification guidance as needed
See NCCN Guidelines® for the complete list of recommendations.
What does this guidance mean for ibrutinib (IMBRUVICA®)?
- NCCN Guidelines recommend continuation of the same cBTKi until progression and/or intolerance2
- ibrutinib (IMBRUVICA®) is the only cBTKi with recommended dose reductions for select ARs* after first occurrence for appropriate patients1
- ibrutinib (IMBRUVICA®) has exploratory outcomes data in patients following dose modification in 1L CLL3
Certain types and severity of cardiac ARs require discontinuations after first occurrence for only ibrutinib (IMBRUVICA®).
NCCN makes no warranties of any kind whatsoever regarding their content, use, or application, and disclaims any responsibility for their application or use in any way.
A peer perspective: NCCN guidance on dose modifications
NCCN makes no warranties of any kind whatsoever regarding their content, use, or application, and disclaims any responsibility for their application or use in any way.
Dose modifications were designed to help manage ARs1
Initial dose modifications for pooled IMBRUVICA®-treated patients (cardiac and non-cardiac related)4
Study Designs
- This analysis pooled data from the IMBRUVICA® arm of two multicenter studies of patients with treatment-naïve CLL/SLL. RESONATE™-2 included 136 patients who were 65 years or older and received single-agent IMBRUVICA®. iLLUMINATE™ included 113 patients who were 65 years or older or with coexisting medical conditions and received IMBRUVICA® in combination with obinutuzumab1
- Median (range) follow-up times varied between studies as follows: 86 months (0-97) for RESONATE™-2 and 43 months (0-52) for iLLUMINATE™4
- Dose modifications were defined in the study protocol; presented with outcomes are only those modified according to current recommendations
Pooled IMBRUVICA®-treated patients: N=248
Patients with any AE leading to a dose reduction per protocol = 48 (19%)
Dose modifications were per USPI and per study protocol.
Patients with an AE leading
to dose reduction per USPI*
21/248 (9%)
AE recurrence
- No recurrence or recurred at lower grade: 16/21 (76%)
- Recurred at the same or higher grade: 5/21 (24%)
Recurrence can take place after AE resolution.
Of the 21 patients who required a dose reduction†‡§
Initial AE resolution:
for pooled IMBRUVICA®-treated patients
95%
20/21
Initial AE that led to a dose modification was resolved in 95% of patients
Timing of AE outcomes was not part of the evaluation.
In patients who both did and did not dose modify over the entire course of the long-term follow-up phase 3 trials
22.9%
57/249
22.9% of patients discontinued IMBRUVICA® due to AEs5
Timing of AE outcomes, including resolution, recurrence, and discontinuation was not part of the evaluation. Dose modifications include a dose hold, followed by a dose reduction.
Results from this analysis are descriptive in nature and have no implications regarding efficacy.
These pooled analysis results are not included in the Prescribing Information for IMBRUVICA®.
From the USPI
- Adverse reactions leading to dose reduction occurred in approximately 9% of patients
- 4% to 10% of patients with CLL/SLL receiving IMBRUVICA® discontinued treatment due to adverse reactions
AEs for which dose reductions are recommended in the USPI (grade 2 cardiac failure, grade 3 cardiac arrhythmia, grade 3-4 non-hematological AEs [excluding cardiac failure and cardiac arrhythmia], grade 3-4 neutropenia with infection or fever, and grade 4 hematological AEs).
†The denominator is patients with AEs for which dose reductions are recommended in the IMBRUVICA® USPI.
‡Two patients had two different AESIs that led to dose reduction; for one patient, neither recurred, and for the other patient, one did not recur and the other (rash maculo-papular) recurred at a lower grade.
§Five patients had AEs that recurred at the same grade (grade 3 atrial fibrillation [n=1], grade 3 diarrhea [n=1], grade 3 headache [n=1], grade 4 neutropenia [n=1], and grade 3 pleural effusion [n=1]); all resolved without further dose reduction.
RESONATE™-2 long-term analysis: Dose modifications can help manage ARs so appropriate patients can continue to benefit1
RESONATE™-2 exploratory post hoc analysis:
PFS in IMBRUVICA®-treated patients with or without dose modifications due to AEs3
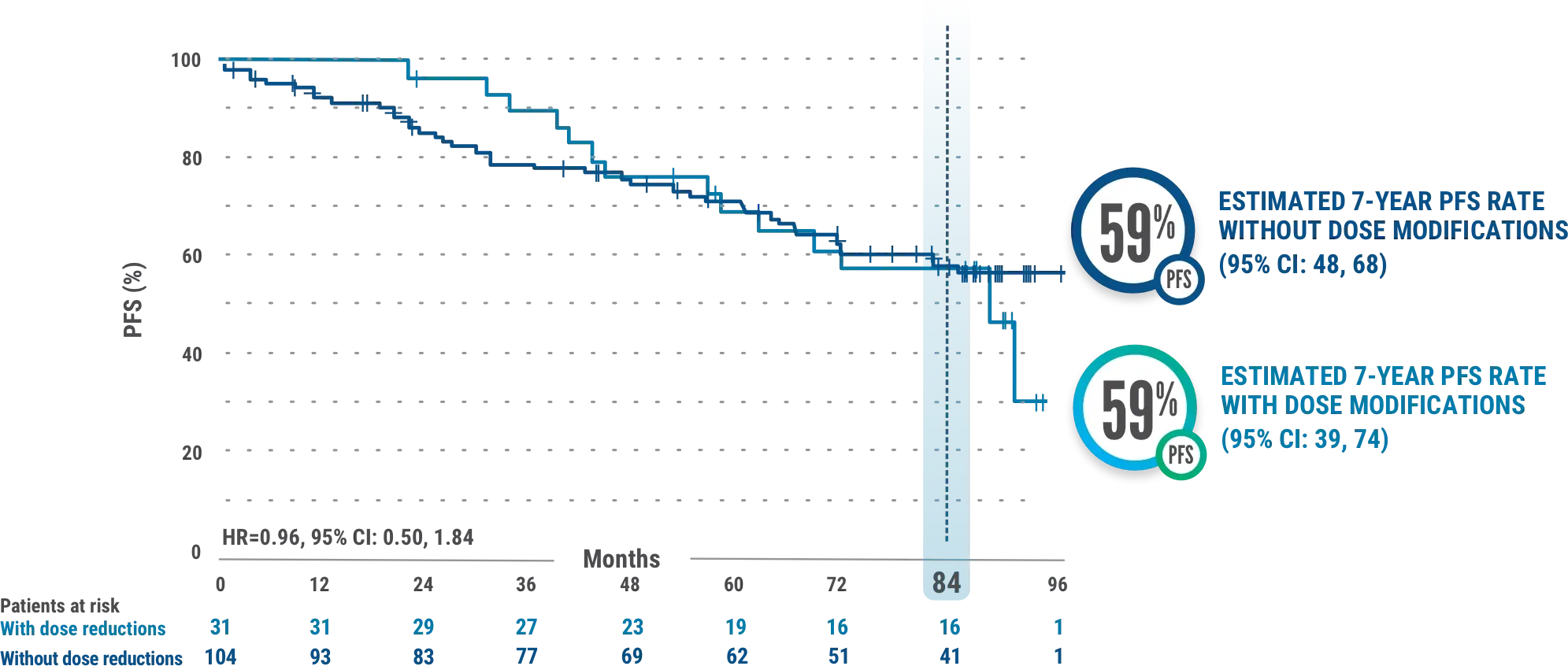
This exploratory analysis evaluated baseline demographics and clinical outcomes (PFS) in subgroups of patients with and without dose reductions due to AEs from the overall population of IMBRUVICA®-treated CLL patients.
Study Context
- The median time to first dose reduction was 23.7 months (range, 1.6-78.4)6
- Median PFS for IMBRUVICA®-treated patients with dose reductions was 87.7 months (95% CI: 56.9-NE) and was not reached (95% CI: 81.9-NE) for those without dose reductions3
- Median follow-up was 82.7 months (range, 0.1-96.6 months). Results from the tail end of the Kaplan-Meier curve should be interpreted with caution due to low number of patients at risk3
- AEs led to dose reductions in 23% (31/135) of all patients treated with IMBRUVICA®3
- The median duration of IMBRUVICA® treatment was 74 months (range, 0.7-96.6 months)7
- Results are based on a starting dosage of IMBRUVICA® 420 mg once daily. Kaplan-Meier curves ≥7.5 years have a limited sample size, potentially impacting PFS estimates3
- Subgroups of patients with and without dose modifications were not stratified for any baseline characteristics. Imbalances in baseline characteristics may exist between these groups3
- Outcomes in the subgroup of patients with and without dose reductions in the overall population of all IMBRUVICA®-treated patients from RESONATE™-2 are from exploratory post hoc analyses and were not powered for significance; comparative statistics are provided for descriptive purposes only3
- Dose modifications for any reason were per protocol based on the discretion of the physician3
8-year RESONATE™-2 long-term dosing results are not included in the Prescribing Information for IMBRUVICA®.

IMBRUVICA® dose modification data reinforces our initial decision to dose modify based on guidance from the Prescribing Information.”
Lisa Nodzon, PhD, ARNP, AOCNP
RESONATE™-2 8-year data: Dose modifications for ARs3
Median overall treatment duration in RESONATE™-27
74
MONTHS
The median duration of IMBRUVICA® treatment was 74 months (range, 0.7-96.6 months; n=135)7
Median treatment duration following dose modifications3
36
MONTHS
In a subset of patients who had dose reductions due to adverse reactions (n=31), the median duration of treatment with IMBRUVICA® after the dose modification was 36.1 months (range, 0.0-84+ months)3*
Study Context
- These data are descriptive in nature only and have no implications regarding efficacy or safety
- Dose modifications were defined in the study protocol; patients may not have undergone ibrutinib dose reduction according to current recommendations
- The data described below reflect exposure to IMBRUVICA® in one single-arm, open-label clinical trial (Study 1102) and 5 randomized controlled clinical trials (RESONATE™, RESONATE™-2, HELIOS, iLLUMINATE™, and E1912) in patients with CLL/SLL (n=2,016 total, including n=1,133 patients exposed to IMBRUVICA®)1
- 4%-10% of patients with CLL/SLL receiving IMBRUVICA® discontinued treatment due to ARs. These included pneumonia, hemorrhage, atrial fibrillation, neutropenia, arthralgia, rash, and thrombocytopenia. ARs leading to ibrutinib dose reduction occurred in approximately 9% of patients1
Duration of dose reduction to study drug discontinuation was calculated from first date of the earliest dose reduction to study drug discontinuation.3
Related resources
Abbreviations
AE=adverse event, AESI=adverse event of special interest, AR=adverse reaction, APP=advanced practice provider, cBTKi=covalent Bruton's tyrosine kinase inhibitor, CI=confidence interval, CLL=chronic lymphocytic leukemia, CYP3A=cytochrome P450, family 3, subfamily A, HR=hazard ratio, NE=not estimable, PFS=progression-free survival, SLL=small lymphocytic lymphoma.
References
1. IMBRUVICA® (ibrutinib) Prescribing Information. 2. Referenced with permission from the NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines®) for Chronic Lymphocytic Leukemia/Small Lymphocytic Lymphoma V.2.2025. © National Comprehensive Cancer Network, Inc. 2025. All rights reserved. Accessed February 24, 2025. To view the most recent and complete version of the guideline, go online to NCCN.org. 3. Woyach JA, Barr PM, Kipps TJ, et al. Characteristics and clinical outcomes of patients with chronic lymphocytic leukemia/small lymphocytic lymphoma receiving ibrutinib for 5 years in the RESONATE-2 Study. Cancers (Basel). 2023;15(2):507. 4. Ghia P, Owen C, Barrientos JC, et al. Initiating first-line ibrutinib in patients with chronic lymphocytic leukemia improves overall survival outcomes to rates approximating an age-matched population of ≥65 years. Poster presented at: 64th Annual Meeting of the American Society of Hematology; December 10-13, 2022; New Orleans, LA. 5. Data on file ABVRRTI75538 6. Data on file ABVRRTI76430 7. Barr PM, Owen C, Robak T, et al. Up to 8-year follow-up from RESONATE-2: first-line ibrutinib treatment for patients with chronic lymphocytic leukemia. Blood Adv. 2022;6(11):3440-3450.